Données techniques
| Formule | C38H38N4O6 |
||||||
| Poids moléculaire | 646.73 | Numéro CAS | 206873-63-4 | ||||
| Solubilité (25°C)* | In vitro | DMSO | 8 mg/mL (12.36 mM) | ||||
| Water | Insoluble | ||||||
| Ethanol | Insoluble | ||||||
| In vivo (Ajouter les solvants au produit individuellement et dans lordre.) |
|
||||||
|
* <1 mg/ml signifie légèrement soluble ou insoluble. * Veuillez noter que Selleck teste la solubilité de tous les composés en interne, et la solubilité réelle peut différer légèrement des valeurs publiées. Ceci est normal et est dû à de légères variations dun lot à lautre. * Expédition à température ambiante (les tests de stabilité montrent que ce produit peut être expédié sans aucune mesure de refroidissement.) |
|||||||
Préparation des solutions mères
Activité biologique
| Description | Tariquidar est un inhibiteur non compétitif puissant et sélectif de la P-glycoprotéine avec un Kd de 5,1 nM dans la lignée cellulaire CHrB30, inversant la résistance aux médicaments dans les lignées cellulaires MDR. Phase 3. | ||
|---|---|---|---|
| Cibles |
|
||
| In vitro | Tariquidar présente une liaison de haute affinité à la P-gp avec un Bmax de 275 pmol/mg. Ce composé montre une interaction non-compétitive avec les substrats de la P-gp. Il augmente l'accumulation à l'état d'équilibre de ces cytotoxiques dans les cellules CHrB30 à des niveaux observés dans les cellules AuxB1 n'exprimant pas de P-gp avec une EC50 de 487 nM. Ce produit chimique est capable d'inhiber l'activité ATPase sensible au vanadate de la P-gp de 60 à 70%, avec de puissantes valeurs d'IC50 de 43 nM. Il peut inhiber d'autres mécanismes de résistance à des concentrations plus élevées. 1 µM de ce composé abroge la résistance médiatisée par l'ABCG2 (BCRP) aux camptothécines in vitro. Il potentialise la cytotoxicité de plusieurs médicaments, y compris la doxorubicine; une inversion complète de la résistance est obtenue en présence de 25 à 80 nM de ce produit chimique. Dans MC26, une lignée cellulaire de carcinome du côlon murin avec une chimiorésistance intrinsèque, l'IC50 de la doxorubicine est cinq fois inférieure en présence de 0,1 µM de ce composé (36 vs 7 nM). Dans les lignées cellulaires de carcinome mammaire murin, de carcinome pulmonaire à petites cellules humain et de carcinome ovarien humain avec une chimiorésistance acquise (EMT6/AR1.0, H69/LX4 et 2780 AD), l'IC50 de la doxorubicine in vitro est 22 à 150 fois inférieure en présence de 0,1 µM de ce produit chimique. L'inhibition de la P-gp persiste pendant 23 h après son retrait du système de culture. Il a restauré la cytotoxicité de la doxorubicine dans le modèle de sphéroïde tumoral multicellulaire NCI/ADRRES (National Cancer Institute) dérivé de la lignée cellulaire de cancer du sein MCF7WT. |
||
| In vivo | Tariquidar (2-8 mg/kg p.o.) s'est avéré potentialiser significativement l'activité antitumorale de la doxorubicine (5 mg/kg, i.v.) contre le carcinome murin du côlon MC26 in vivo. Dans les xénogreffes de carcinome humain, la coadministration de ce composé (6-12 mg/kg p.o.) a entièrement restauré l'activité antitumorale contre deux xénogreffes tumorales humaines MDR hautement résistantes (2780AD, H69/LX4) chez des souris nues. |
Protocole (de référence)
| Test kinase : |
|
|---|---|
| Test cellulaire : |
|
| Étude animale : |
|
Références
|
Validation du produit par le client
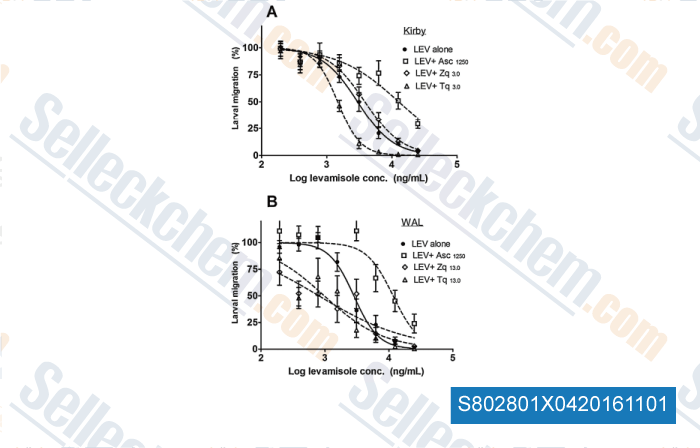
-
, , Vet Parasitol, 2015, 211(1-2):80-8.
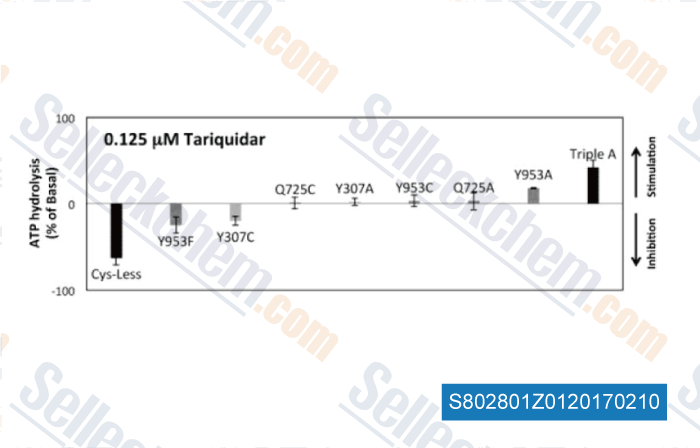
-
Données de [ , , Biochem Pharmacol, 2016, 101:40-53. ]
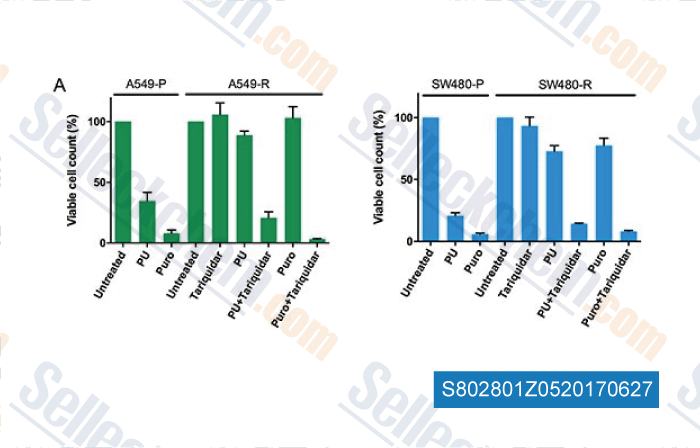
-
Données de [ , , Oncotarget, 2017, 8(5):7678-7690 ]
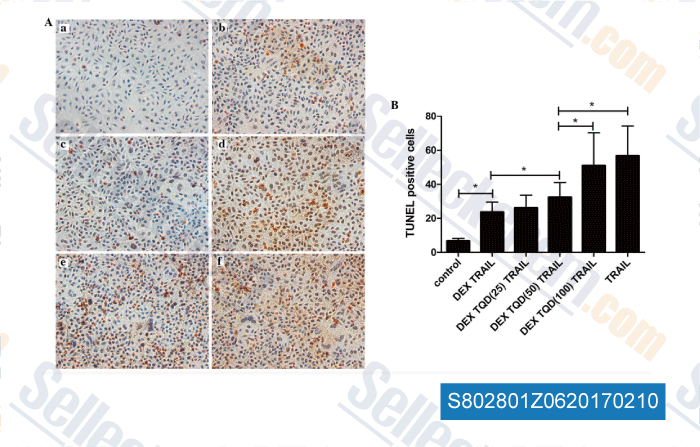
-
Données de [ , , Mol Med Rep, 2015, 12(6):8093-100. ]
Sellecks Tariquidar (XR9576) A été cité par 55 Publications
| Identification of the Notch ligand DLK1 as an immunotherapeutic target and regulator of tumor cell plasticity and chemoresistance in adrenocortical carcinoma [ Nat Commun, 2025, 16(1):5511] | PubMed: 40595495 |
| Mitochondrial dysfunction fuels drug resistance in adult T-cell acute lymphoblastic leukemia [ J Transl Med, 2025, 23(1):542] | PubMed: 40369632 |
| Fibroblasts Attenuate Anti-Tumor Drug Efficacy in Tumor Cells via Paracrine Interactions with Tumor Cells in 3D Plexiform Neurofibroma Cultures [ Cells, 2025, 14(16)1276] | PubMed: 40862755 |
| Low-Dose Perifosine, a Phase II Phospholipid Akt Inhibitor, Selectively Sensitizes Drug-Resistant ABCB1-Overexpressing Cancer Cells [ Biomol Ther (Seoul), 2025, 33(1):170-181] | PubMed: 39632683 |
| Tumor-acquired somatic mutation affects conformation to abolish ABCG2-mediated drug resistance [ Drug Resist Updat, 2024, 73:101066] | PubMed: 38387283 |
| Enhanced anticancer effect of thymidylate synthase dimer disrupters by promoting intracellular accumulation [ Front Pharmacol, 2024, 15:1477318] | PubMed: 39611169 |
| Zosuquidar: An Effective Molecule for Intracellular Ca2+ Measurement in P-gp Positive Cells [ Int J Mol Sci, 2024, 25(6)3107] | PubMed: 38542082 |
| Cellular uptake of CPX-351 by scavenger receptor class B type 1-mediated nonendocytic pathway [ Exp Hematol, 2024, S0301-472X(24)00516-2] | PubMed: 39362576 |
| Ivermectin Enhances Paclitaxel Efficacy by Overcoming Resistance Through Modulation of ABCB1 in Non-small Cell Lung Cancer [ Anticancer Res, 2024, 44(12):5271-5282] | PubMed: 39626921 |
| The net electrostatic potential and hydration of ABCG2 affect substrate transport [ Nat Commun, 2023, 14(1):5035] | PubMed: 37596258 |
POLITIQUE DE RETOUR
La politique de retour inconditionnelle de Selleck Chemical garantit une expérience dachat en ligne fluide à nos clients. Si vous nêtes en aucun cas satisfait de votre achat, vous pouvez retourner tout article dans les 7 jours suivant sa réception. En cas de problèmes de qualité du produit, quil sagisse de problèmes liés au protocole ou au produit, vous pouvez retourner tout article dans les 365 jours suivant la date dachat initiale. Veuillez suivre les instructions ci-dessous lors du retour des produits.
EXPÉDITION ET STOCKAGE
Les produits Selleck sont transportés à température ambiante. Si vous recevez le produit à température ambiante, soyez assuré que le service dinspection de la qualité de Selleck a mené des expériences pour vérifier que le placement à température normale pendant un mois naffectera pas lactivité biologique des produits en poudre. Après réception, veuillez stocker le produit conformément aux exigences décrites dans la fiche technique. La plupart des produits Selleck sont stables dans les conditions recommandées.
NON DESTINÉ À UN USAGE HUMAIN, VÉTÉRINAIRE DIAGNOSTIQUE OU THÉRAPEUTIQUE.