pour la recherche uniquement
Alvimopan Opioid Receptor antagoniste
Réf. CatalogueS5935
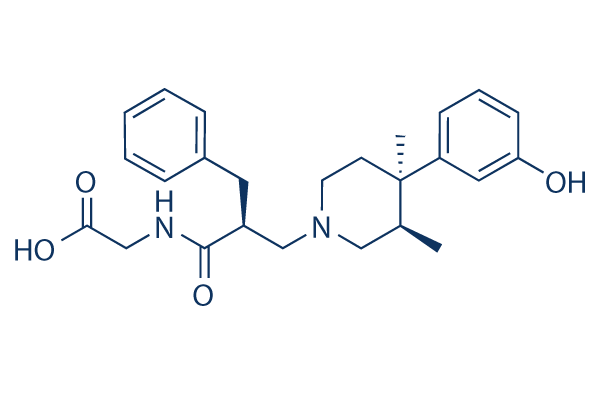
Structure chimique
Poids moléculaire: 424.53
Contrôle Qualité
| Cibles apparentées | Adrenergic Receptor AChR 5-HT Receptor COX Calcium Channel Histamine Receptor Dopamine Receptor GABA Receptor TRP Channel Cholinesterase (ChE) |
|---|---|
| Autre Opioid Receptor Inhibiteurs | JTC-801 (+)-Matrine Racecadotril Trimebutine Docusate Sodium Trimebutine maleate Asimadoline hydrochloride BMS-986122 Naloxegol Oxalate methylnaltrexone bromide |
Informations chimiques, stockage et stabilité
| Poids moléculaire | 424.53 | Formule | C25H32N2O4 |
Stockage (À compter de la date de réception) | 3 years -20°C powder |
|---|---|---|---|---|---|
| N° CAS | 156053-89-3 | -- | Stockage des solutions mères |
|
|
| Synonymes | LY 246736, ADL 8-2698 | Smiles | CC1CN(CCC1(C)C2=CC(=CC=C2)O)CC(CC3=CC=CC=C3)C(=O)NCC(=O)O | ||
Solubilité
|
In vitro |
DMSO
: 85 mg/mL
(200.22 mM)
Water : Insoluble Ethanol : Insoluble |
Calculateur de molarité
|
In vivo |
|||||
Calculateur de formulation in vivo (Solution claire)
Étape 1 : Saisir les informations ci-dessous (Recommandé : Un animal supplémentaire pour tenir compte des pertes pendant lexpérience)
Étape 2 : Saisir la formulation in vivo (Ceci est seulement le calculateur, pas la formulation. Veuillez nous contacter dabord sil ny a pas de formulation in vivo dans la section Solubilité.)
Résultats du calcul :
Concentration de travail : mg/ml;
Méthode de préparation du liquide maître DMSO : mg médicament prédissous dans μL DMSO ( Concentration du liquide maître mg/mL, Veuillez nous contacter dabord si la concentration dépasse la solubilité du DMSO du lot de médicament. )
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, puis ajouterμL PEG300, mélanger et clarifier, puis ajouterμL Tween 80, mélanger et clarifier, puis ajouter μL ddH2O, mélanger et clarifier.
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, puis ajouter μL Huile de maïs, mélanger et clarifier.
Note : 1. Veuillez vous assurer que le liquide est clair avant dajouter le solvant suivant.
2. Assurez-vous dajouter le(s) solvant(s) dans lordre. Vous devez vous assurer que la solution obtenue, lors de lajout précédent, est une solution claire avant de procéder à lajout du solvant suivant. Des méthodes physiques telles que le vortex, les ultrasons ou le bain-marie chaud peuvent être utilisées pour faciliter la dissolution.
Mécanisme daction
| Targets/IC50/Ki |
μ-opioid receptor
(Cell-free assay) 0.77 nM(Ki)
δ-opioid receptor
(Cell-free assay) 4.4 nM
δ-opioid receptor
(Cell-free assay) 4.4 nM(Ki)
κ-opioid receptor
(Cell-free assay) 40 nM(Ki)
|
|---|---|
| In vitro |
L'Alvimopan est hautement sélectif (par ≥227 fois) pour le récepteur ", humain par rapport au sous-type ",, mais présente une sélectivité plus modeste (≥6 fois) pour le récepteur ",/". Dans l'iléon isolé de cobaye, ce composé est un puissant antagoniste de l'inhibition des contractions évoquées électriquement, induite par la morphine, le DAMGO ou l'endomorpine-1, et médiée par le ", Opioid Receptor (valeurs de pA2 de 9,6 ou 9,7). Les puissances antagonistes ", et ", de cette substance chimique sont plus faibles dans l'iléon de cobaye (valeurs de pA2 de 8,7 et 7,8, respectivement). Ce composé (1 ou 10 ",M) n'a pas d'affinité significative pour un large éventail de récepteurs non opioïdes, de canaux ioniques et d'enzymes pour lesquels il a été testé. |
| In vivo |
Chez les animaux, l'alvimopan antagonise l'analgésie induite par la morphine, médiatisée de manière centrale, uniquement à des doses relativement élevées, avec des concentrations plasmatiques très élevées nécessaires pour traverser la barrière hémato-encéphalique. Après administration intraveineuse, ce composé est environ 200 fois plus puissant pour bloquer les récepteurs ", périphériques que centraux. Après administration orale, il est également très actif. Chez le chien, l'administration intraveineuse de cette substance chimique a entraîné des augmentations dose-dépendantes des concentrations plasmatiques maximales et de l'aire plasmatique sous la courbe de concentration-temps. Cependant, en raison d'une mauvaise absorption systémique, des doses orales allant jusqu'à 100 mg/kg ont produit de faibles concentrations plasmatiques (Cmax moyenne = 92,9 ng/ml), ce qui a entraîné une biodisponibilité orale d'environ 0,03 %. La demi-vie de cet agent est estimée à environ 10 minutes après administration intraveineuse chez les chiens et les lapins. |
Références |
|
Informations sur lessai clinique
(données du https://clinicaltrials.gov, mis à jour le 2024-05-22)
| Numéro NCT | Recrutement | Conditions | Promoteur/Collaborateurs | Date de début | Phases |
|---|---|---|---|---|---|
| NCT03068975 | Terminated | Post Operative Ileus |
University of Puerto Rico|Merck Sharp & Dohme LLC |
September 1 2017 | Phase 4 |
| NCT00259922 | Completed | Bowel Dysfunction|Constipation |
Cubist Pharmaceuticals LLC a subsidiary of Merck & Co. Inc. (Rahway New Jersey USA)|GlaxoSmithKline |
August 2005 | Phase 3 |
| NCT00256932 | Completed | Bowel Dysfunction|Constipation |
Cubist Pharmaceuticals LLC a subsidiary of Merck & Co. Inc. (Rahway New Jersey USA)|GlaxoSmithKline |
August 2005 | Phase 3 |
| NCT00241722 | Completed | Bowel Dysfunction|Constipation |
Cubist Pharmaceuticals LLC a subsidiary of Merck & Co. Inc. (Rahway New Jersey USA)|GlaxoSmithKline |
August 1 2005 | Phase 3 |
Support technique
Tel: +1-832-582-8158 Ext:3
Si vous avez dautres questions, veuillez laisser un message.
Les produits sont destinés à la recherche uniquement. Ne pas utiliser chez lhomme. Nous ne vendons pas aux patients.
©Copyright 2013 Selleck Chemicals. Tous droits réservés.