pour la recherche uniquement
Fimasartan Angiotensin Receptor antagoniste
Réf. CatalogueS4975
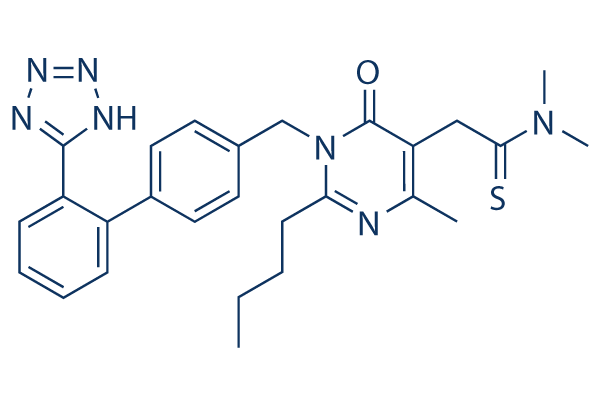
Structure chimique
Poids moléculaire: 501.65
Contrôle Qualité
- Cité dans Nature Medicine pour sa qualité supérieure
- COA
- SDS
- Fiche technique
| Cibles apparentées | CXCR Hedgehog/Smoothened PKA Adrenergic Receptor AChR 5-HT Receptor Histamine Receptor Dopamine Receptor Ras KRas |
|---|---|
| Autre Angiotensin Receptor Inhibiteurs | PD123319 ML221 A-779 Olodanrigan (EMA401) Buloxibutid AVE 0991 |
Informations chimiques, stockage et stabilité
| Poids moléculaire | 501.65 | Formule | C27H31N7OS |
Stockage (À compter de la date de réception) | |
|---|---|---|---|---|---|
| N° CAS | 247257-48-3 | -- | Stockage des solutions mères |
|
|
| Synonymes | Kanarb | Smiles | CCCCC1=NC(=C(C(=O)N1CC2=CC=C(C=C2)C3=CC=CC=C3C4=NNN=N4)CC(=S)N(C)C)C | ||
Solubilité
|
In vitro |
DMSO
: 100 mg/mL
(199.34 mM)
Ethanol : 24 mg/mL Water : Insoluble |
Calculateur de molarité
|
In vivo |
|||||
Calculateur de formulation in vivo (Solution claire)
Étape 1 : Saisir les informations ci-dessous (Recommandé : Un animal supplémentaire pour tenir compte des pertes pendant lexpérience)
Étape 2 : Saisir la formulation in vivo (Ceci est seulement le calculateur, pas la formulation. Veuillez nous contacter dabord sil ny a pas de formulation in vivo dans la section Solubilité.)
Résultats du calcul :
Concentration de travail : mg/ml;
Méthode de préparation du liquide maître DMSO : mg médicament prédissous dans μL DMSO ( Concentration du liquide maître mg/mL, Veuillez nous contacter dabord si la concentration dépasse la solubilité du DMSO du lot de médicament. )
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, puis ajouterμL PEG300, mélanger et clarifier, puis ajouterμL Tween 80, mélanger et clarifier, puis ajouter μL ddH2O, mélanger et clarifier.
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, puis ajouter μL Huile de maïs, mélanger et clarifier.
Note : 1. Veuillez vous assurer que le liquide est clair avant dajouter le solvant suivant.
2. Assurez-vous dajouter le(s) solvant(s) dans lordre. Vous devez vous assurer que la solution obtenue, lors de lajout précédent, est une solution claire avant de procéder à lajout du solvant suivant. Des méthodes physiques telles que le vortex, les ultrasons ou le bain-marie chaud peuvent être utilisées pour faciliter la dissolution.
Mécanisme daction
| Targets/IC50/Ki |
AT1 receptor
|
|---|---|
| In vitro |
Le Fimasartan est un antagoniste sélectif du récepteur AT1. La concentration qui inhibe la liaison de [125I]Ang II au récepteur AT1 de la corticosurrénale de rat de 50 % (IC50) est de 0,13 nM. Ce composé atténue puissamment la mort apoptotique myocardique dans les cœurs de rat reperfusés et les cellules H9c2. Il pourrait atténuer les dommages mitochondriaux associés à la minimisation de la réduction de Bcl-2 et liés à la réduction de p53 et à l'activation d'Akt et de GSK-3β, et inhiber la surcharge mitochondriale en Ca2+ en supprimant l'ICa,L et MCU. |
| In vivo |
Dans divers modèles animaux, y compris les rats hypertendus rénaux, les rats spontanément hypertendus et les chiens Beagle, le fimasartan réduit efficacement la pression artérielle de manière dose-dépendante après administration orale et intraveineuse unique ou répétée. Ce composé n'affecte pas le comportement général, la fréquence respiratoire ou le volume courant chez les animaux de laboratoire, et ne présente aucun effet indésirable dans le test hERG (human ether-a-go-go-related gene) ou l'étude télémétrique chez le singe. Un certain nombre d'études de toxicité générale, de toxicité cancérogène, de toxicité génétique et de toxicité sur le développement, administrées par voie orale ou intraveineuse chez diverses espèces, y compris les souris, les rats, les singes et les chiens, ont été menées et ces résultats précliniques démontrent la sécurité et la tolérabilité de cette substance chimique pour une utilisation clinique à long terme et offrent des marges de sécurité suffisantes pour soutenir la dose thérapeutique humaine attendue. Il est rapidement absorbé après administration orale, le temps nécessaire pour atteindre la concentration plasmatique maximale (Tmax) variant de 0,5 à 3 h et la demi-vie terminale (t1/2) étant de 5 à 16 h à des doses de 20 à 480 mg chez des sujets sains. Des résultats similaires sont obtenus chez les patients atteints d'hypertension, c'est-à-dire que le Tmax varie de 0,5 à 1,3 h et la t1/2 est de 7 à 10 h après l'administration de ce composé à des doses de 20 à 180 mg dans l'étude de phase II ultérieure. L'excrétion urinaire de cette substance chimique est faible, l'excrétion urinaire totale du médicament inchangé au cours des 24 premières heures après l'administration étant inférieure à 3 % de la dose administrée. Il subit une élimination non rénale avec un métabolisme minimal. Le traitement avec ce composé avant l'ischémie myocardique atténue significativement les lésions de reperfusion et les changements apoptotiques, possiblement en supprimant les dommages mitochondriaux pendant la reperfusion. Dans diverses études précliniques, y compris les modèles de lésion par ballonnet aortique, d'ischémie/reperfusion d'infarctus du myocarde, de cardiotoxicité de la doxorubicine et d'accident vasculaire cérébral ischémique, cette substance chimique présente des effets anti-inflammatoires et protecteurs des organes. |
Références |
|
Informations sur lessai clinique
(données du https://clinicaltrials.gov, mis à jour le 2024-05-22)
| Numéro NCT | Recrutement | Conditions | Promoteur/Collaborateurs | Date de début | Phases |
|---|---|---|---|---|---|
| NCT02994745 | Completed | Hypertension Hyperlipidemia |
Boryung Pharmaceutical Co. Ltd |
December 23 2016 | Phase 1 |
| NCT02920047 | Completed | Hypertension |
Boryung Pharmaceutical Co. Ltd |
October 2016 | Phase 1 |
| NCT02995720 | Completed | Hypertension Hyperlipidemia |
Boryung Pharmaceutical Co. Ltd |
August 26 2016 | Phase 1 |
| NCT03231293 | Completed | Hypertension|Ischemic Stroke|Transient Ischemic Attack |
Boryung Pharmaceutical Co. Ltd |
July 28 2016 | -- |
Support technique
Tel: +1-832-582-8158 Ext:3
Si vous avez dautres questions, veuillez laisser un message.
Les produits sont destinés à la recherche uniquement. Ne pas utiliser chez lhomme. Nous ne vendons pas aux patients.
©Copyright 2013 Selleck Chemicals. Tous droits réservés.