pour la recherche uniquement
PRN1371 FGFR inhibiteur
Réf. CatalogueS8578
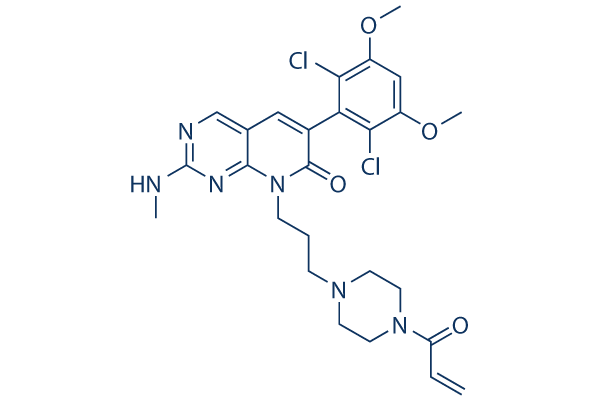
Structure chimique
Poids moléculaire: 561.46
Contrôle Qualité
| Cibles apparentées | EGFR VEGFR JAK PDGFR Src HIF FLT FLT3 HER2 Bcr-Abl |
|---|---|
| Autre FGFR Inhibiteurs | PD173074 AZD4547 (Fexagratinib) BLU9931 Futibatinib (TAS-120) LY2874455 PD-166866 Zoligratinib (Debio-1347) H3B-6527 Fisogatinib (BLU-554) SSR128129E |
Informations chimiques, stockage et stabilité
| Poids moléculaire | 561.46 | Formule | C26H30Cl2N6O4 |
Stockage (À compter de la date de réception) | |
|---|---|---|---|---|---|
| N° CAS | 1802929-43-6 | Télécharger le SDF | Stockage des solutions mères |
|
|
| Synonymes | N/A | Smiles | CNC1=NC=C2C=C(C(=O)N(C2=N1)CCCN3CCN(CC3)C(=O)C=C)C4=C(C(=CC(=C4Cl)OC)OC)Cl | ||
Solubilité
|
In vitro |
DMSO
: 56 mg/mL
(99.73 mM)
Water : Insoluble Ethanol : Insoluble |
Calculateur de molarité
|
In vivo |
|||||
Calculateur de formulation in vivo (Solution claire)
Étape 1 : Saisir les informations ci-dessous (Recommandé : Un animal supplémentaire pour tenir compte des pertes pendant lexpérience)
Étape 2 : Saisir la formulation in vivo (Ceci est seulement le calculateur, pas la formulation. Veuillez nous contacter dabord sil ny a pas de formulation in vivo dans la section Solubilité.)
Résultats du calcul :
Concentration de travail : mg/ml;
Méthode de préparation du liquide maître DMSO : mg médicament prédissous dans μL DMSO ( Concentration du liquide maître mg/mL, Veuillez nous contacter dabord si la concentration dépasse la solubilité du DMSO du lot de médicament. )
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, puis ajouterμL PEG300, mélanger et clarifier, puis ajouterμL Tween 80, mélanger et clarifier, puis ajouter μL ddH2O, mélanger et clarifier.
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, puis ajouter μL Huile de maïs, mélanger et clarifier.
Note : 1. Veuillez vous assurer que le liquide est clair avant dajouter le solvant suivant.
2. Assurez-vous dajouter le(s) solvant(s) dans lordre. Vous devez vous assurer que la solution obtenue, lors de lajout précédent, est une solution claire avant de procéder à lajout du solvant suivant. Des méthodes physiques telles que le vortex, les ultrasons ou le bain-marie chaud peuvent être utilisées pour faciliter la dissolution.
Mécanisme daction
| Targets/IC50/Ki |
FGFR1
(Cell-free assay) 0.6 nM
FGFR2
(Cell-free assay) 1.3 nM
FGFR3
(Cell-free assay) 4.1 nM
CSF1R
(Cell-free assay) 8.1 nM
FGFR4
(Cell-free assay) 19.3 nM
|
|---|---|
| In vitro |
PRN1371 est un inhibiteur nanomolaire irréversible de FGFR1-4. Ce composé présente un profil unique de haute puissance biochimique et cellulaire (FGFR1 IC50 = 0,6 nM, SNU16 IC50 = 2,6 nM), un engagement prolongé de la cible (occupation de FGFR1 24 h = 96 %), une inhibition du hERG <30 % à 1 μM et une bonne stabilité ADME prédite avec une réactivité BME Kd>100 μM. Cette substance chimique a maintenu une occupation élevée de FGFR1 avec une solubilité améliorée et une biodisponibilité orale exceptionnelle.
|
| In vivo |
Une étude PK chez le rat iv (2 mg/kg) de PRN1371 a montré une clairance rapide (Cl = 160 ml/min/kg), mais l'administration po (20 mg/kg) a démontré une exposition orale élevée (AUC = 4348 h·ng/mL) et une demi-vie raisonnable (t1/2 = 3,8 h). Les études PK de ce composé chez le rat, le chien et le singe cynomolgus ont montré une clairance iv rapide chez toutes les espèces ; cependant, il y avait de grandes différences interspécifiques dans l'exposition orale et la biodisponibilité pour le singe par rapport au rat et au chien. Chez le rat, une exposition élevée après administration orale (par exemple, Cmax = 1785 ng/mL, AUC = 4348 ng·h/mL) et une biodisponibilité (F) >100 % suggéraient une bonne absorption et une saturation partielle des mécanismes de clairance à la dose de 20 mg/kg. Spécifique au rat, il existe une grande différence de demi-vie entre les voies d'administration iv (t1/2 = 0,8 h) et po (t1/2 = 3,8 h), ce qui indique également une saturation possible d'un mécanisme de clairance lors de l'administration orale. Chez les chiens, la même formulation de suspension de méthylcellulose utilisée pour le rat a donné une faible absorption orale et une faible biodisponibilité (F < 15 %). Dans le modèle de xénogreffe de cancer gastrique SNU16 chez la souris, ce composé a induit une réduction dose-dépendante du volume tumoral et jusqu'à 68 % d'inhibition de la croissance tumorale à la dose la plus élevée de 10 mg/kg deux fois par jour après 27 jours de traitement. Toutes les doses ont été bien tolérées sans perte de poids corporel significative.
|
Références |
Applications
| Méthodes | Biomarqueurs | Images | PMID |
|---|---|---|---|
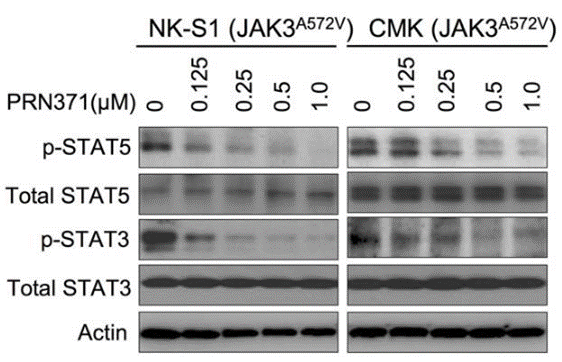
| Western blot | p-STAT5 / STAT5 / p-STAT3 / STAT3 |
|
29434279 |
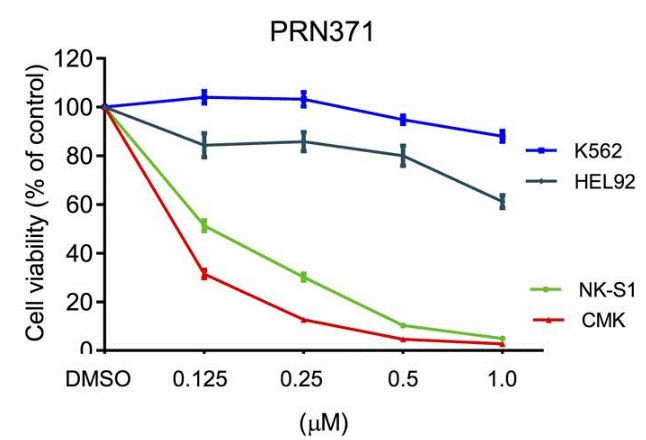
| Growth inhibition assay | Cell viability |
|
29434279 |
Informations sur lessai clinique
(données du https://clinicaltrials.gov, mis à jour le 2024-05-22)
| Numéro NCT | Recrutement | Conditions | Promoteur/Collaborateurs | Date de début | Phases |
|---|---|---|---|---|---|
| NCT02608125 | Terminated | Solid Tumors|Metastatic Urothelial Carcinoma & Renal Pelvis & Ureter |
Principia Biopharma a Sanofi Company |
October 28 2015 | Phase 1 |
Support technique
Tel: +1-832-582-8158 Ext:3
Si vous avez dautres questions, veuillez laisser un message.
Les produits sont destinés à la recherche uniquement. Ne pas utiliser chez lhomme. Nous ne vendons pas aux patients.
©Copyright 2013 Selleck Chemicals. Tous droits réservés.