pour la recherche uniquement
Idarubicin HCl Topoisomerase inhibiteur
Réf. CatalogueS1228
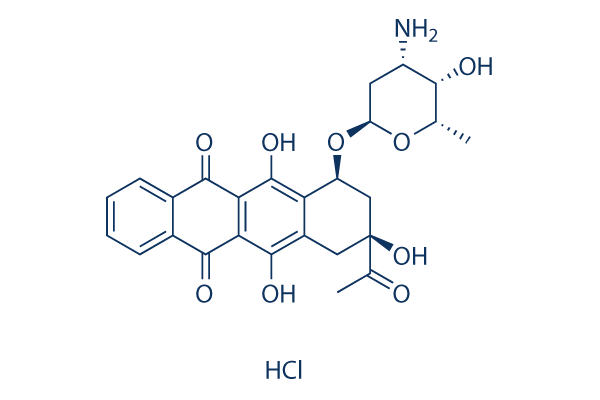
Structure chimique
Poids moléculaire: 533.95
Contrôle Qualité
| Cibles apparentées | HDAC PARP ATM/ATR DNA-PK WRN DNA/RNA Synthesis PPAR Sirtuin Casein Kinase eIF |
|---|---|
| Autre Topoisomerase Inhibiteurs | Camptothecin (CPT) Betulinic acid Beta-Lapachone (S)-10-Hydroxycamptothecin Amonafide Voreloxin (SNS-595) hydrochloride Ellagic acid Cu(II)-Elesclomol Hydroxy Camptothecine Rubitecan |
Culture cellulaire, traitement et concentration de travail
| Lignées cellulaires | Type dessai | Concentration | Temps dincubation | Formulation | Description de lactivité | PMID |
|---|---|---|---|---|---|---|
| K562 erythroleukemic cells | Cytotoxic assay | Cytotoxic activity against K562 erythroleukemic cells, IC50=0.002 μM | 10425093 | |||
| K562 cells | Cytotoxic assay | 5 days | Cytotoxicity against human K562 cells after 5 days by XTT assay, IC50=0.012 μM | 18076140 | ||
| mouse DA-3 cells | Cytotoxic assay | 24 h | Cytotoxicity against mouse DA-3 cells after 24 hrs by XTT assay, IC50=2.3 μM | 24900668 | ||
| human ES2 cells | Cytotoxic assay | 24 h | Cytotoxicity against human ES2 cells after 24 hrs by XTT assay, IC50=3.2 μM | 24900668 | ||
| human K562 cells | Proliferation assay | 72 h | Antiproliferative activity against human K562 cells after 72 hrs, GI50=3.3 μM | 25420175 | ||
| human SKOV3 cells | Cytotoxic assay | 24 h | Cytotoxicity against human SKOV3 cells after 24 hrs by XTT assay, IC50=4.5 μM | 24900668 | ||
| HepG2 cells | Function assay | Inhibition of liver stage Plasmodium berghei infection in HepG2 cells, IC50=6.62 μM | 22586124 | |||
| human MCF7 cells | Cytotoxic assay | 24 h | Cytotoxicity against human MCF7 cells after 24 hrs by XTT assay, IC50=7.3 μM | 24900668 | ||
| mouse DA-3 cells | Cytotoxic assay | 20 μM | Cytotoxicity against mouse DA-3 cells assessed as reduction in cell viability at 20 uM by XTT assay | 24900668 | ||
| neural precursor cells | Function assay | Inhibition of neurosphere proliferation of mouse neural precursor cells by MTT assay | 17417631 | |||
| VERO-E6 | Function assay | 48 hrs | Toxicity CC50 against VERO-E6 cells determined at 48 hours by high content imaging (same conditions as 2_LEY without exposure to 0.01 MOI SARS CoV-2 virus), CC50=0.2μM | ChEMBL | ||
| Cliquez pour voir plus de données expérimentales sur la lignée cellulaire | ||||||
Informations chimiques, stockage et stabilité
| Poids moléculaire | 533.95 | Formule | C26H27NO9.HCl |
Stockage (À compter de la date de réception) | |
|---|---|---|---|---|---|
| N° CAS | 57852-57-0 | Télécharger le SDF | Stockage des solutions mères |
|
|
| Synonymes | 4-demethoxydaunorubicin (NSC256439, 4-DMDR) HCl | Smiles | CC1C(C(CC(O1)OC2CC(CC3=C2C(=C4C(=C3O)C(=O)C5=CC=CC=C5C4=O)O)(C(=O)C)O)N)O.Cl | ||
Solubilité
|
In vitro |
DMSO
: 100 mg/mL
(187.28 mM)
Water : 8 mg/mL Ethanol : Insoluble |
Calculateur de molarité
|
In vivo |
|||||
Calculateur de formulation in vivo (Solution claire)
Étape 1 : Saisir les informations ci-dessous (Recommandé : Un animal supplémentaire pour tenir compte des pertes pendant lexpérience)
Étape 2 : Saisir la formulation in vivo (Ceci est seulement le calculateur, pas la formulation. Veuillez nous contacter dabord sil ny a pas de formulation in vivo dans la section Solubilité.)
Résultats du calcul :
Concentration de travail : mg/ml;
Méthode de préparation du liquide maître DMSO : mg médicament prédissous dans μL DMSO ( Concentration du liquide maître mg/mL, Veuillez nous contacter dabord si la concentration dépasse la solubilité du DMSO du lot de médicament. )
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, puis ajouterμL PEG300, mélanger et clarifier, puis ajouterμL Tween 80, mélanger et clarifier, puis ajouter μL ddH2O, mélanger et clarifier.
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, puis ajouter μL Huile de maïs, mélanger et clarifier.
Note : 1. Veuillez vous assurer que le liquide est clair avant dajouter le solvant suivant.
2. Assurez-vous dajouter le(s) solvant(s) dans lordre. Vous devez vous assurer que la solution obtenue, lors de lajout précédent, est une solution claire avant de procéder à lajout du solvant suivant. Des méthodes physiques telles que le vortex, les ultrasons ou le bain-marie chaud peuvent être utilisées pour faciliter la dissolution.
Mécanisme daction
| Caractéristiques |
Idarubicin is a substrate for CYP450 2D6 and 2C9.
|
|---|---|
| Targets/IC50/Ki |
Topo II (MCF-7 cells)
(Cell-free assay) 3.3 ng/mL
Multicellular spheroids
(Cell-free assay) 7.9 ng/mL
|
| In vitro |
Idarubicin a une activité cytotoxique significative contre les sphéroïdes multicellulaires, comparable aux effets antiprolifératifs sur les cellules en monocouche. Idarubicin inhibe le CYP450 2D6. Idarubicin est environ 57,5 fois et 25 fois plus active que la doxorubicine et l'épirubicine, respectivement. Idarubicin est capable de surmonter la résistance multidrogue médiatisée par la P-glycoprotéine. Idarubicin inhibe la formation de radicaux superoxydes par les PMN. Idarubicin pourrait être couplée aux anticorps monoclonaux (anti-Ly-2.1, anti-L3T4 ou anti-Thy-1) avec rétention de la solubilité des protéines et de l'activité des anticorps. Idarubicin inhibe la prolifération des cellules NALM-6 avec une IC50 de 12 nM. |
| Essai kinase |
Expériences de métabolisme du CYP450
|
|
L'évaluation du métabolisme d'Idarubicin par les isoenzymes du CYP450 3A4, 2D6, 2C8, 2C9 et 1A2 est réalisée à l'aide de protéines CYP450 humaines isolées pour chaque isoforme. La méthode de test d'inhibition du P450 à haut débit est utilisée pour ces évaluations. Les expériences de métabolisme sont conçues pour étudier les propriétés suivantes de chaque médicament : (1) si Idarubicin est un substrat des isoenzymes CYP450 3A4, 2C8, 2C9, 1A2 ou 2D6 ; (2) si le métabolisme est affecté par des inhibiteurs connus de chaque isoenzyme ; (3) si Idarubicin est un inhibiteur des isoenzymes CYP450 ; et (4) si la caspofungine ou l'itraconazole inhibent le métabolisme du CYP450 d'Idarubicin. Le dibenzylfluorésceine (DBF) (CYP3A4, CYP2C8, CYP2C9), le 3-cyano-7-éthoxycoumarine (Cyp1A2) et le 7-méthoxy-4-(aminométhyl)-coumarine (MAMC) (CYP2D6) sont les substrats connus utilisés comme contrôles pour confirmer l'activité de l'isoenzyme respective et évaluer les effets d'Idarubicin sur l'activité de l'isoenzyme. De plus, le kétoconazole, la quercétine, le suflaphenazole, la furafylline et la quinidine sont utilisés comme inhibiteurs de contrôle du CYP450 pour les isoenzymes 3A4, 2C8, 2C9, 1A2 ou 2D6, respectivement. Le substrat, l'inhibiteur plus Idarubicin tel qu'indiqué sont ajoutés à chaque échantillon de protéine et incubés pendant 20 à 60 minutes, comme recommandé par le fabricant, à 37 °C. Les réactions sont arrêtées avec une solution de solvant organique, puis les échantillons sont analysés par lecteur de plaque à fluorescence, le cas échéant. Pour chaque expérience, des échantillons de contrôle avec une quantité connue de substrat et de métabolite synthétisé, en l'absence de l'isoenzyme, sont préparés pour des comparaisons qualitatives. Toutes les expériences sont réalisées en triple.
|
|
| In vivo |
La réduction d'Idarubicin est dépendante des cétone réductases et procède plus stéréosélectivement que celle de la plupart des cétones, donnant presque exclusivement l'épimère (13S). La haute stéréospécificité de la réduction d'Idarubicin pourrait résulter de l'induction chirale due à la présence de centres asymétriques près du groupe carbonyle dans Idarubicin. |
Références |
|
Informations sur lessai clinique
(données du https://clinicaltrials.gov, mis à jour le 2024-05-22)
| Numéro NCT | Recrutement | Conditions | Promoteur/Collaborateurs | Date de début | Phases |
|---|---|---|---|---|---|
| NCT05697510 | Recruiting | Acute Myeloid Leukemia (AML) |
Nantes University Hospital |
March 16 2023 | Phase 1 |
| NCT02652871 | Completed | Leukemia |
M.D. Anderson Cancer Center|Eli Lilly and Company|High Impact Clinical Research Support Program |
May 9 2016 | Phase 1 |
Support technique
Tel: +1-832-582-8158 Ext:3
Si vous avez dautres questions, veuillez laisser un message.
Les produits sont destinés à la recherche uniquement. Ne pas utiliser chez lhomme. Nous ne vendons pas aux patients.
©Copyright 2013 Selleck Chemicals. Tous droits réservés.