pour la recherche uniquement
Fenebrutinib (GDC-0853) BTK inhibiteur
Réf. CatalogueS8421
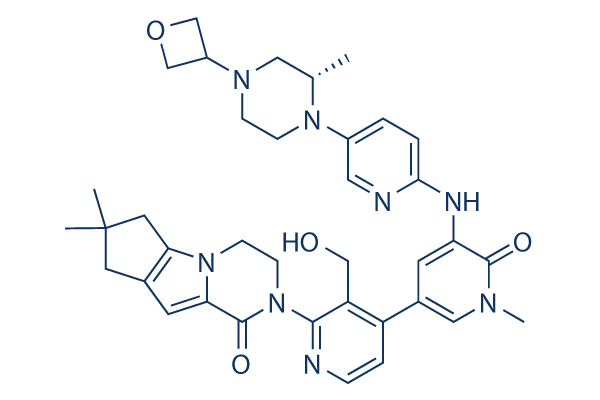
Structure chimique
Poids moléculaire: 664.80
Contrôle Qualité
- Cité dans Nature Medicine pour sa qualité supérieure
- COA
- NMR
- SDS
- Fiche technique
| Cibles apparentées | EGFR VEGFR PDGFR FGFR c-Met Src MEK CSF-1R FLT3 HER2 |
|---|---|
| Autre BTK Inhibiteurs | Catadegbrutinib (BGB-16673) Spebrutinib (AVL-292) tirabrutinib(ONO-4059) hydrochloride CGI1746 LFM-A13 CNX-774 Evobrutinib BMS-935177 Branebrutinib (BMS-986195) Pirtobrutinib (LOXO-305) |
Informations chimiques, stockage et stabilité
| Poids moléculaire | 664.80 | Formule | C37H44N8O4 |
Stockage (À compter de la date de réception) | 3 years -20°C powder |
|---|---|---|---|---|---|
| N° CAS | 1434048-34-6 | -- | Stockage des solutions mères |
|
|
| Synonymes | N/A | Smiles | CC1CN(CCN1C2=CN=C(C=C2)NC3=CC(=CN(C3=O)C)C4=C(C(=NC=C4)N5CCN6C7=C(CC(C7)(C)C)C=C6C5=O)CO)C8COC8 | ||
Solubilité
|
In vitro |
DMSO
: 8 mg/mL
(12.03 mM)
Water : Insoluble Ethanol : Insoluble |
Calculateur de molarité
|
In vivo |
|||||
Calculateur de formulation in vivo (Solution claire)
Étape 1 : Saisir les informations ci-dessous (Recommandé : Un animal supplémentaire pour tenir compte des pertes pendant lexpérience)
Étape 2 : Saisir la formulation in vivo (Ceci est seulement le calculateur, pas la formulation. Veuillez nous contacter dabord sil ny a pas de formulation in vivo dans la section Solubilité.)
Résultats du calcul :
Concentration de travail : mg/ml;
Méthode de préparation du liquide maître DMSO : mg médicament prédissous dans μL DMSO ( Concentration du liquide maître mg/mL, Veuillez nous contacter dabord si la concentration dépasse la solubilité du DMSO du lot de médicament. )
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, puis ajouterμL PEG300, mélanger et clarifier, puis ajouterμL Tween 80, mélanger et clarifier, puis ajouter μL ddH2O, mélanger et clarifier.
Méthode de préparation de la formulation in vivo : Prendre μL DMSO liquide maître, puis ajouter μL Huile de maïs, mélanger et clarifier.
Note : 1. Veuillez vous assurer que le liquide est clair avant dajouter le solvant suivant.
2. Assurez-vous dajouter le(s) solvant(s) dans lordre. Vous devez vous assurer que la solution obtenue, lors de lajout précédent, est une solution claire avant de procéder à lajout du solvant suivant. Des méthodes physiques telles que le vortex, les ultrasons ou le bain-marie chaud peuvent être utilisées pour faciliter la dissolution.
Mécanisme daction
| Targets/IC50/Ki |
BTK
(Cell-free assay) 0.91 nM(Ki)
BTK C481R
(Cell-free assay) 1.3 nM(Ki)
BTK C481S
(Cell-free assay) 1.6 nM(Ki)
BTK T474M
(Cell-free assay) 3.4 nM(Ki)
BTK T474I
(Cell-free assay) 12.6 nM(Ki)
|
|---|---|
| In vitro |
Testé à 1 μM contre un large panel d'essais biochimiques de kinases humaines, le Fenebrutinib (GDC-0853) n'inhibe que 3 des 286 kinases hors cible. Basé sur les valeurs d'IC50 déterminées, la sélectivité pour la Btk est >100 fois supérieure à chacune de ces 3 cibles secondaires : Bmx (153 fois), Fgr (168 fois) et Src (131 fois). Ce composé bloque à la fois la signalisation BCR des lymphocytes B et la signalisation FcγR des monocytes. Dans un essai enzymatique biochimique de Btk in vitro, il présente un temps de résidence moyen avec Btk de 18,3 ± 2,8 heures. Il bloque l'autophosphorylation cellulaire de la Btk de type sauvage (WT Btk) et du mutant C481S. Les cellules de LLC (leucémie lymphoïde chronique) traitées avec le GDC-0853 in vitro avant la stimulation du BCR montrent des niveaux réduits de phosphorylation de la BTK et une activation diminuée des cibles en aval, notamment PLCγ2, AKT et ERK. Il inhibe la transcription dépendante du NF-κB, réduit l'activation et altère la migration. Ce composé n'inhibe pas l'EGFR et l'ITK dans le système cellulaire et n'affecte pas l'activation du récepteur des lymphocytes T. |
| Essai kinase |
Sélectivité de la kinase
|
|
La sélectivité de la kinase Fenebrutinib (GDC-0853) est évaluée à une concentration de 1 µM dans un panel allant jusqu'à 287 essais d'activité et de liaison de kinases humaines recombinantes, y compris les tyrosine kinases cytoplasmiques et réceptrices, les sérine/thréonine kinases et les lipid kinases. Les essais d'activité kinase mesurent la phosphorylation des peptides ou la production d'ADP tandis que les essais de liaison surveillent le déplacement des sondes de liaison au site ATP. Les concentrations d'ATP utilisées dans les essais d'activité sont généralement dans un facteur de 2 de la valeur de la constante de Michaelis apparente (Kmapp) déterminée expérimentalement pour chaque kinase, tandis que les concentrations de traceur de liaison compétitif utilisées dans les essais de liaison sont généralement dans un facteur de 3 des valeurs de la constante de dissociation (Kd) déterminées expérimentalement. Ce composé est testé en double contre chaque kinase et les valeurs moyennes de % d'inhibition sont rapportées. Pour les kinases qui sont inhibées à près ou plus de 80 % à la concentration testée, des titrations d'inhibiteurs en 10 points utilisant les mêmes essais sont effectuées afin de déterminer les concentrations d'inhibiteurs qui ont provoqué 50 % d'inhibition (IC50).
|
|
| In vivo |
Le Fenebrutinib (GDC-0853) a une clairance modérée de 27,4 mL/min/kg et une excellente biodisponibilité (F=65%) chez les rats ayant reçu 0,2 mg/kg par injection intrapéritonéale ou 1 mg/kg PO. Sa clairance plasmatique est de 27,4 mL/min/kg, le volume de distribution (Vd) est de 5,42 L/kg et la demi-vie plasmatique (t1/2) est de 2,2 h. Ce composé présente également des propriétés PK favorables chez le chien. La demi-vie de 3,8 heures (Clp 10,9 mL/min/kg, Vd 2,96 L/kg) et la biodisponibilité orale élevée (85%) permettent également d'atteindre des expositions suffisantes dans les études toxicologiques chez le chien. Il est bien toléré chez les rats et les chiens et présente un profil de sécurité globalement favorable. Le GDC-0853 est utile dans le traitement de la polyarthrite rhumatoïde et d'autres maladies auto-immunes médiées par les cellules B ou myéloïdes. Dans une étude à dose unique croissante (SAD) (0,5 mg à 600 mg) et une étude à doses multiples croissantes (MAD) pendant 14 jours (250 mg BID à 500 mg QD), il est très bien toléré sans événements indésirables graves, sans signaux de sécurité et sans toxicités limitant la dose. Il est bien absorbé et présente une pharmacocinétique linéaire et proportionnelle à la dose. Chez les rats Sprague-Dawley (SD), l'administration de ce composé et d'autres inhibiteurs de la BTK structurellement diversifiés pendant 7 jours ou plus provoque des lésions pancréatiques consistant en une hémorragie multifocale centrée sur les îlots, une inflammation, une fibrose et des macrophages chargés de pigment avec une atrophie, une dégénérescence et une inflammation des cellules acineuses exocrines lobulaires adjacentes. Des résultats similaires ne sont pas observés chez les souris ou les chiens à des expositions beaucoup plus élevées. |
Références |
|
Applications
| Méthodes | Biomarqueurs | Images | PMID |
|---|---|---|---|
| Western blot | p-BTK / BTK / p-PLCγ2 / PLCγ2 / p-AKT / AKT / p-ERK / ERK |
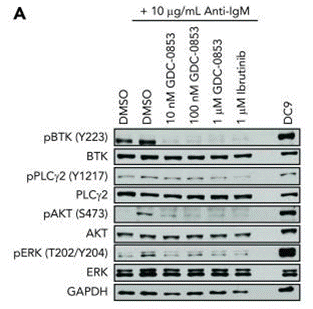
|
30018078 |
Informations sur lessai clinique
(données du https://clinicaltrials.gov, mis à jour le 2024-05-22)
| Numéro NCT | Recrutement | Conditions | Promoteur/Collaborateurs | Date de début | Phases |
|---|---|---|---|---|---|
| NCT05119569 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 1 2022 | Phase 2 |
| NCT04586023 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 24 2021 | Phase 3 |
| NCT04586010 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 17 2021 | Phase 3 |
| NCT03693625 | Terminated | Urticaria |
Genentech Inc. |
September 27 2018 | Phase 2 |
| NCT03596632 | Completed | Healthy Participants |
Hoffmann-La Roche |
July 27 2018 | Phase 1 |
Support technique
Tel: +1-832-582-8158 Ext:3
Si vous avez dautres questions, veuillez laisser un message.
Les produits sont destinés à la recherche uniquement. Ne pas utiliser chez lhomme. Nous ne vendons pas aux patients.
©Copyright 2013 Selleck Chemicals. Tous droits réservés.